The orbitals can be plotted using two types of outputs :
Molden files recommended
Cube files
Molden files can be created from orca output by using the orca_2mkl tool.
Cube files can be created from orca output by using the orca_plot tool.
Using molden files instead of cube files allows to calculate the atomic orbitals (AO) and the molecular orbitals (MO) on-the-fly on a grid the user can choose. This also enables the calculation of all the MO using a single molden file instead of multiple cube files.
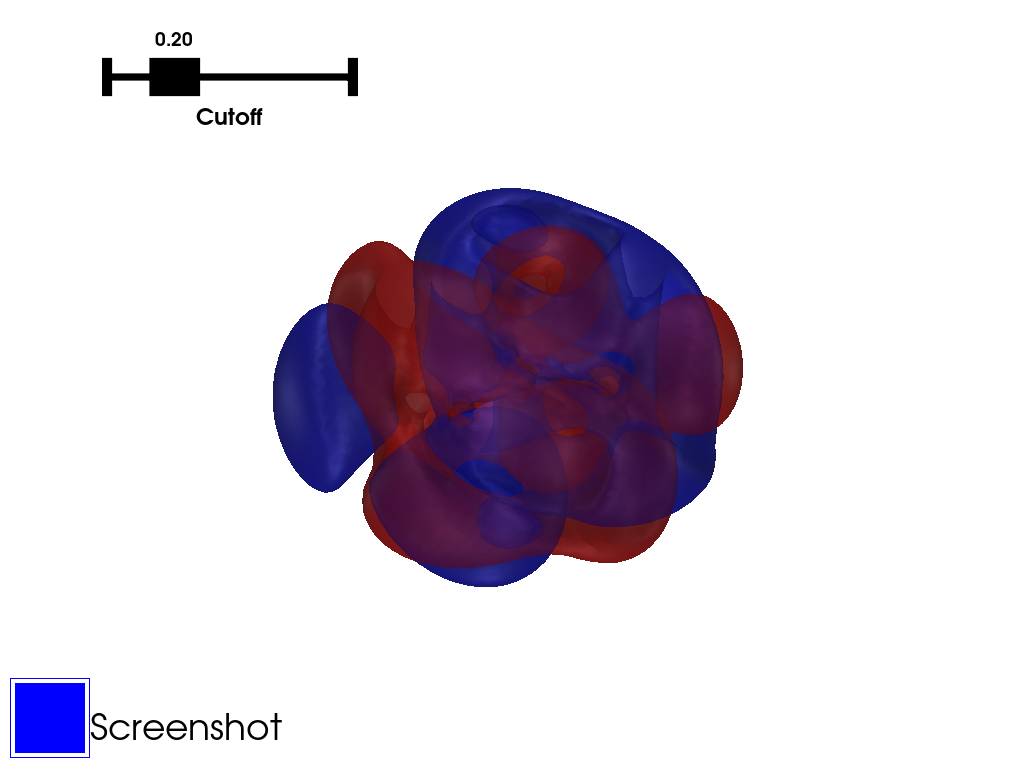
With the sum being on all the transitions of the excitation, \(Occ_i(r)\) and \(Virt_i(r)\) being respectively the occupied and the virtual molecular orbitals considered in the transition, and \(c_i\) the coefficient of the transition.
The transition densities can therefore be computed using the MO calculated from molden file combined with the extraction of the coefficients from a TD-DFT calculation.
If one wants to use Orca for the TD-DFT calculation, triplets states should not be included and the keyword TPRINT 0.00001 will allow more transitions to be printed.
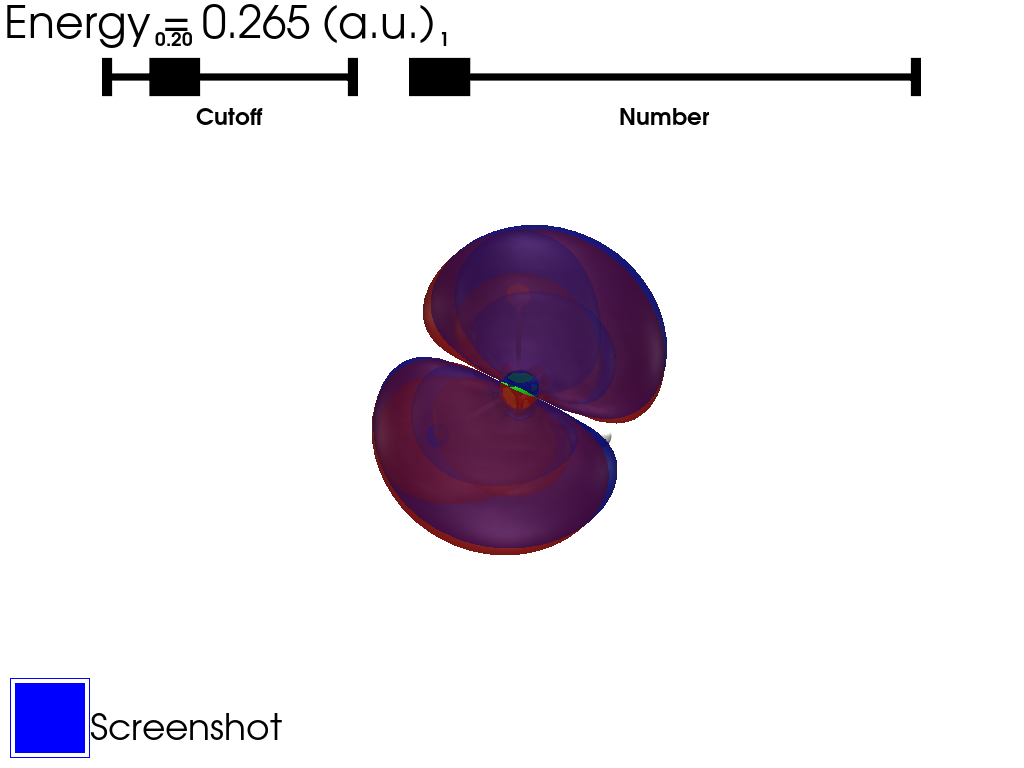
They are sorted by energy. They can be selected using a slider which cannot be accessed using the interactive scene. A script contaning all the examples can be found here.